
O E M E R G O
August 2016 a UL company
Picture a scenario where you are busily answering emails and then the phone rings. The person on the other line is with the local Food and Drug Administration office, letting you know that
they will be at your facility on Monday morning. It is Wednesday afternoon, giving you two business days to make sure your company is fully compliant. If your company is outside of the United States, usually you have a bit more time, as the visit needs to be planned in advance for travel logistics.

NOTICE OF INSPECTION
Sincerely,
WINFLO | MEDICAL DEVICES
Regardless, now your organization is facing one of the most dreaded events that a medical device
company must endure… an FDA Quality System Regulation (QSR) inspection. This paper will discuss the activities leading up to the inspection, what to expect during an inspection, and what companies should do after the FDA investigator leaves the facility. There are many horror stories associated with an FDA inspection, but hopefully this discussion will dispel many of those fears and help your organization prepare for an FDA QSR inspection.
The first aspect we should discuss is the actual set of requirements a company would be expected to comply with regarding manufacturing and distributing a finished medical device. The primary regulation that must be followed is the Code of Federal Regulations Title 21 CFR 820, Quality System Regulations (QSR)1. However, it should be noted that there are a number of other regulations that must be followed and are often forgotten as manufacturers are concentrating on QSR requirements. These include the following:
21 CFR 801, Labeling
21 CFR 803, Medical Device Reporting
21 CFR 806, Reports of Corrections and Removals
21 CFR 807, Establishment Registration and Device Listing
21 CFR 810, Medical Device Recall Authority
and the newest one 21 CFR 830, Unique Device Identification
All of these regulations must be followed as applicable to the organization. When the phrase “as applicable” is mentioned, this relates to the fact that throughout the regulations there may be requirements that are not applicable or appropriate for an organization. As an example, if an organization distributes a single-use device, then servicing of the device, i.e. 21 CFR 820.200, would not be applicable. Another example is if an organization has a Class I device not requiring
design controls, then 21 CFR 820.30 would not be applicable to that manufacturer. It is important that organizations understand application of the FDA regulations and compliance expectations when adhering to these regulations.
The FDA developed a methodology for performing inspections; this was developed specifically by Office of Regulatory Affairs (ORA), the group within the agency responsible for conducting inspections of medical device manufacturers. This guide, called the Quality System Inspection Technique or QSIT2, was developed during release of the QSR regulations in 1996.

FDA Inspection
Procedures Winf lo | Medi ca l Devices
The QSIT guide- which is to be used in conjunction with the various compliance program guides, regulations, and guidance documents- provides
a foundation for performing QSR inspections.
FDA designed the QSIT as a “top down” approach; it focuses on the organization’s entire quality system to identify potential quality issues within the company.
The QSIT focuses on four major subsystems of the quality system shown below that establish the basis for an FDA investigator to review procedures, processes, and quality records. The FDA published the QSIT guide to demonstrate what they will review during an
inspection; therefore, organizations should read and study the QSIT guide and apply its principles when they review and audit their quality system.
FDA QSR Subsystems |
1. Management Controls: Quality Manual, Quality Policy, Quality Planning, Management Commitment, Management Reviews, Training and Personnel, Internal Audits |
2. Corrective and Preventative Action (CAPA): Sources of Data, Nonconformities, Customer Complaints, Medical Device Reporting, Follow-up Activities |
3. Design Controls: Design Planning, Design Inputs, Design Outputs, Verification, Validation, Risk Management, Design Reviews, Device Master Record |
4. Production and Process Controls: Product Realization (Device Master Record), Manufacturing, Process Validation, Calibration, Identification, Traceability, Device History Record, Labeling, Packaging, Storage, Delivery, Servicing |
One of the most effective ways to ensure a successful FDA inspection is to always be “inspection ready.” Even though the FDA generally pre-announces inspections, a few days or even a few weeks are not enough time to correct fundamental compliance issues in a quality system. This is the reason why a medical device manufacturer should always be inspection-ready, which follows the main theme that procedures have been established and successfully implemented, records are being maintained to show conformance and, when applicable, that issues identified are being corrected.

The primary method for accomplishing this is to have a robust internal audit program. Emergo frequently advises organizations to utilize their internal audit process to locate quality system problems first rather than letting FDA inspectors find them. Ensure that the internal audit program is unbiased, and that personnel in the organization are forthcoming when describing their processes. The worst cases arise when personnel hide issues from their internal auditor because they do not want to get reprimanded. This helps no one when the FDA identifies these issues during an inspection. Mock audits that mimic FDA inspections can help uncover fundamental compliance issues.
Another good method is to have your quality system periodically reviewed by an independent third party, as internal auditors may get too “close” to their own quality system. As Benjamin Franklin said, “By failing to prepare, you are preparing to fail.” The best method for a successful inspection is to always be prepared.
Assuming your quality system is in good shape and your internal audit procedure is followed closely, you may be wondering what to do once you receive the first call from the FDA. You may not have much time to prepare for the investigator’s arrival, but don’t panic. Refer to the US FDA Audit Checklist at the end of this paper for a step-by-step pre-audit checklist.

When the FDA investigator enters the facility, ensure that the company designates an Inspection Coordinator or an individual to escort FDA personnel, guide the inspection, answer the main questions. This individual should be knowledgeable about the organization. Usually there is only one FDA investigator. If two or more investigators show up, it may be in response to a significant safety-related incident. The FDA investigator should be shown to a segregated location, such as a conference room that can be “home base” for the duration of the inspection. Introductions are made with the FDA investigator stating the purpose of the inspection, which in most cases is a routine QSR inspection. During the opening meeting, this is the opportunity for the organization to provide an overview of the company and its operations.
Once the introductions have been made and background information on your organization has been described, the FDA investigator gets right to business by requesting documentation, commonly referred to as objective evidence. The QSIT method being a “top down” approach
means the inspector generally will ask for procedures first, and then proceed into closer detailed review of quality records. These quality records must support the manufacturer's ongoing activities support how the procedures are written, and show that the processes have been implemented. The bulk of the inspection activity involves the investigator asking for a procedure, interviewing personnel, asking questions about the process, and reviewing records that are generated across all facets of the quality system.
If this is a full QSIT inspection, the investigator will generally cover each of the four major subsystems of the QSIT, one per day. An example of inspection flow is shown in Figure 1. It is important to understand that for domestic inspections (those performed in the United States) an FDA investigator can extend the time required for the inspection if they observe quality issues that need further investigation. It is in your best interest to prepare for a successful inspection so the FDA is not at your facility for two weeks or more, which can be disruptive for the entire organization.
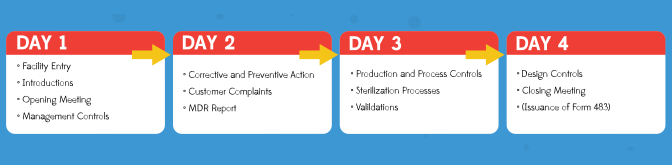
Figure 1: Basic flow of an FDA inspection over four days – note this is just an example as each investigator may shift the subsystems around or take more time per subsystem.
As with any audit or inspection, the FDA inspector must collect a sampling of documents and records, as they are not able to review absolutely everything. This is another reason to be prepared for a FDA inspection as the organization should expect that the FDA investigator would review many procedures and pull numerous quality records that must be readily available.
To prepare for an inspection, your internal audit program should challenge the documentation system, ensuring documents can be provided in a timely manner.

The FDA investigator should follow the process from the QSIT guide, but if they discover quality issues during the inspection this may lead to much more detailed review of specific aspects of the product quality and/or the quality system.
The organization needs to ensure that they document all aspects of the FDA inspection, such as the questions that are asked, the procedures reviewed, and the specific quality records that are examined, etc., so this information can be referenced later or after the inspection. Assign a
note taker or scribe during the FDA inspection who can record the information so the Inspection Coordinator can concentrate on the inspection.
The FDA investigator is a normal person, like you and me; they struggle with time management, have different personalities, or may also experience having a “bad day.” Understand the temperament of the investigator, the lines of questioning, and certainly inquire about their experience or knowledge with certain subjects, e.g. their formal education or years of conducting inspections with the FDA. The organization’s Inspection Coordinator must manage all aspects
of the inspection, anticipate the needs of the investigator, and be present during personnel interviews. Any difficulties experienced during the inspection should be brought to the executive management and/or president immediately, including legal counsel, as needed.
Most Frequent Form 483 Observations |
Corrective and Preventive Action, 21 CFR 820.100: Lack of/inadequate procedures
Customer Complaints, 21 CFR 820.198: Lack of/ inadequate procedures
Purchasing Controls, 21 CFR 820.50: Lack of/ inadequate procedures
Process Validation, 21 CFR 820.75: Lack of/ inadequate validations
Medical Device Reporting, 21 CFR 803: Lack of/ written MDR procedures
The FDA investigator’s purpose is to assess compliance to the QSRs or objectionable practices due to not following Good Manufacturing Practices (GMP); these are the fundamental compliance requirements for medical device manufacturers. FDA publishes metrics identifying problem areas of quality system compliance annually on their website or on public forums like the Regulatory Affairs Professional Society (RAPS). The list in the side bar sums up areas that continually receive the most observations during FDA inspections3 as listed on the FDA Form 483.
Medical device manufacturers can expect that these areas are continually reviewed with more scrutiny by the FDA. This should prepare an organization for concentrating its efforts
and resources on ensuring that these processes are fully compliant within the organization. If there have been quality system/product issues identified in the past, an organization must review these areas specifically to ensure a successful FDA inspection.
The FDA conducts a closing meeting with the organization that summarizes the inspection findings that may or may not result in issuance of an FDA Form 483. FDA Form 483 is an official listing of objectionable findings made during the inspection. The list of observations will be reviewed in the closing meeting
by the FDA investigator to make sure there is no clarification required, and to give the company an opportunity to discuss the Form 483 Observations as necessary.
There is no obligation by the medical device manufacturer to state their intention to correct anything or agree to a certain timeline. The observation(s) can be annotated to state the company promises to correct as part of their response. Assure during the closing meeting that there is no confusion for observation(s) issued and that each one is clearly understood by the organization and the executive management team.
It is also possible not to receive a Form 483! There are a number of medical device manufacturers that have gone through an inspection with no observations being issued; that is a proud achievement to any audit or inspection performed.
Steps to address FDA Inspection observations |
Correction: Actions taken immediately to correct an observation
Root cause analysis: Identify the true root cause; utilize root cause analysis tools like the 5 Why’s
Retrospective review: Review the activities not just related to the observation, but what is being done to review information in the past
Corrective action plan: Actual corrective action that is short or long term to remove the nonconforming situation
Effectiveness check: Describe how the organization will assure the correction and corrective action is sufficient
Timeline: When all of the activities are to be completed
If the organization has received a FDA Form 483 with objectionable items listed, it is imperative that a response is sent to FDA within 15 calendar days. The FDA has often commented in public forums that if a response is not received within that timeframe, then a Warning Letter will be issued. In addition, the response to the observations stated in Form 483 needs to be clearly addressed in detail describing the overall plan for correcting the items.
A basic structure of the Form 483 response is shown in the sidebar, including the content currently expected by the FDA. Deficiencies in the response including missing information, not providing supporting records, or lacking
true commitment to making the corrections may result in a Warning Letter. This is a serious concern because Warning Letters are publicly posted on the FDA website4, allowing your customers and, more importantly—your competitors, to see deficiencies in your quality system.
There is usually no negotiating the content of a Form 483 or Warning Letter after it has been issued, so it is quite important for an organization to stay continually vigilant in the compliance of their quality system.
Be sure to respond quickly to the FDA Form 483 with sufficient information and adequate detail to prevent issuance of a Warning Letter.
After approximately 60 days, the organization should request through the Freedom of Information Act5 to receive a copy of the Establishment Inspection Report (EIR). This is a narrative by the FDA investigator that details what was reviewed, persons interviewed, and comments made during the inspection. This should always be obtained after an inspection is performed to understand any implications or significant comments made regarding an observation by the investigator.

Inform your entire staff that an audit is coming and the dates the FDA inspector will be on-site. Assure that the key individuals are available during the FDA inspection or identified as designated individuals.
If you already have a procedure for handling FDA inspections, your employees should be familiar with the inspection process. However, provide them with a review of the external audit procedure to avoid any surprises.
Designate a space on-site for the investigator to conduct the inspection, such as a large office or conference room. Consider a secondary room as a “back-room” for preparing material for the investigator.
Identify a member of your staff to be the FDA’s point of contact while they are on-site. This person should be a senior member of your quality team so they are familiar with your organization and able to answer main questions.
Executives and senior managers should arrange their schedules to be on-site and available during the audit. Auditors do not like to be kept waiting and it could be detrimental if a member of your senior staff is not available to interview with the investigator.
Your documentation should already be prepared and organized, so this should be easy. However, you should ensure files and documents related to the FDA QSR subsystems are readily accessible to be provided to the investigator in a timely manner.
Alert department representatives and SMEs that they may be interviewed during the audit. Provide any coaching or mentoring for how to interact with the investigator.
Mock interviews should be part of your internal audit procedure, but take the time to review interviewing techniques that will allow you to complete a successful inspection.
This paper provides a brief discussion about what to expect before, during, and after a FDA QSR inspection. However, there are many things that can and do occur during an inspection that cannot be fully detailed in this paper. The important thing to remember is that your organization must always be prepared for an inspection because the FDA can visit your facility at any time.
Utilize the internal audit program and corrective action system to ensure that deficiencies are identified internally, corrected, and support compliance with the requirements. A well-prepared organization can face the challenge of an FDA QSR inspection with the ultimate goal of not receiving any FDA Form 483 observations and knowing they are providing safe, quality products to their customers.

Emergo helps medical device companies with regulatory compliance and market access in the United States and other markets worldwide.
US FDA 510(k) preparation
FDA QSR 21 CFR 820 implementation and training
US FDA Agent Representation
To learn more, visit:
www.emergogroup.com/services/united-states
If you enjoyed this white paper, we know you will like our video explaining how to prepare for a random FDA inspection. We will discuss first steps, important interview strategies, employee involvement, tips and tricks, and much more.
Watch video now See all video content
![]()
References:
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?CFRPart=820
http://www.fda.gov/downloads/iceci/inspections/ucm142981.pdf
The Top 15 Medical Device Deficiencies Cited by FDA in 2014, Regulatory Affairs Professional Society, by Alexander Gaffney, Posted 8 December 2014, Accessed 20 May 2015
http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/default.htm